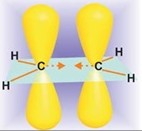
Окисление алкенов (ациклических и циклических) при взаимодействии с перкислотами (надкислотами) в неполярной, индифферентной среде сопровождается образованием окисей алкенов – эпоксиды, поэтому сама реакция носит название реакции эпоксидирования.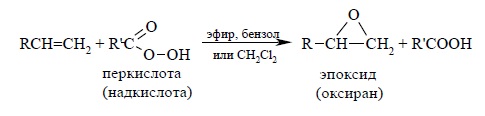

Согласно современно номенклатуре ИЮПАК, трехчленный цикл с одним атомом кислорода носит название оксиран.
Эпоксидирование алкенов следует рассматривать как синхронный, согласованный процесс, в котором не участвуют ионные интермедиаты типа гидроксильного катиона OH+. Эпоксидирование алкенов представляет собой процесс син-присоединения одного атома кислорода по двойной связи с полным сохранением конфигурации заместителей при двойной связи: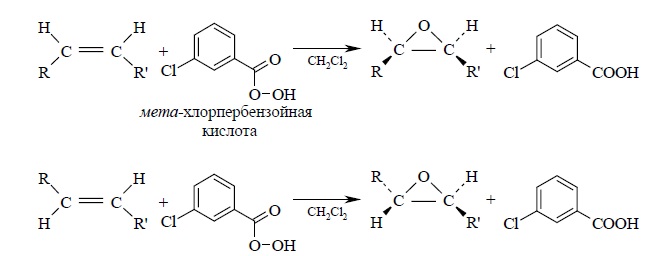

Для эпоксидирования был предложен механизм, характерный для согласованных процессов: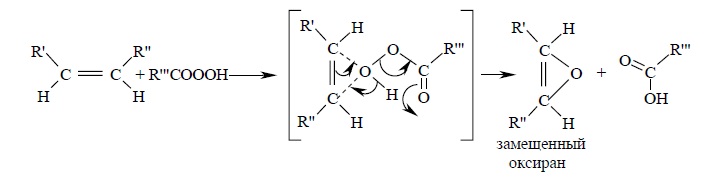

В качестве эпоксидирующих агентов используются перкислоты: пербензойная, м-хлорпербензойная, мононадфталевая, перуксусная, пертрифторуксусная и пермуравьиная. Перкислоты ароматического ряда применяют в виде индивидуальных реагентов, тогда как перкислоты алифатического ряда – CH3CO3H, CF3CO3H и HCO3H – не выделяют индивидуально и используют сразу после их образования при взаимодействии 30- или 90%-й перекиси водорода и соответствующей карбоновой кислоты. Пербензойную и мета-хлорпербензойную кислоты в настоящее время получают окислением соответственно бензойной и мета-хлорбензойной кислот 70%-й перкисью водорода в растворе метансульфокислоты: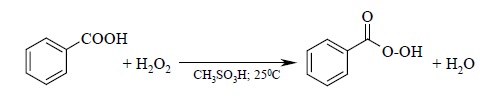

или из хлорангидридов кислот и перекиси водорода: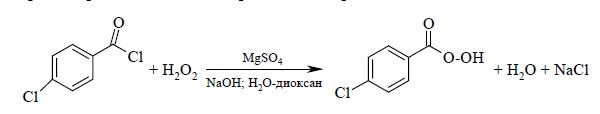

Мононадфталевую кислоту получают подобным методом из фталевого ангидрида и 30%-й перекиси водорода в водной щелочи: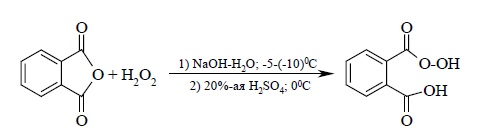
Первоначально для получения оксиранов (эпоксидов) использовалась пербензойная или мононадфталевая кислоты: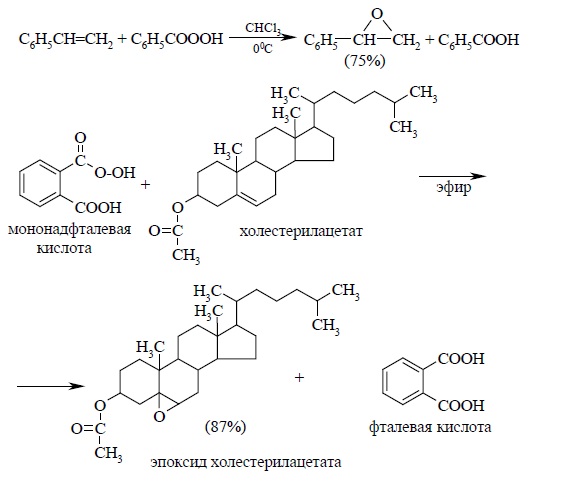

Особенно удобен метод с использованием мононадфталевой кислоты. Мононадфталевая кислота хорошо растворима в эфире, тогда как один из продуктов реакции (фталевая кислота) совершенно не растворим в эфире, и о ходе реакции легко судить по количеству выделившейся кристаллической фталевой кислоты.
В настоящее время для эпоксидирования чаще всего используют мета-хлорпербензойную кислоту. В отличие от других перкислот она стабильная при хранении в течение длительного времени (до 1 года) и абсолютно безопасная при обращении. Выходы оксиранов, полученных при окислении ациклических и циклических алкенов мета-хлорпербензойной кислотой в растворе хлористого метилена, обычно очень высокие.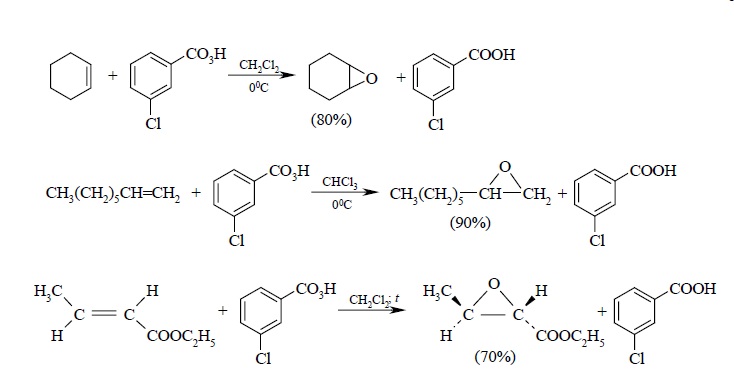

Перкислоты часто генерируют прямо в реакционной смеси из 90%-й перекиси водорода и карбоновой кислоты в хлористом метилене: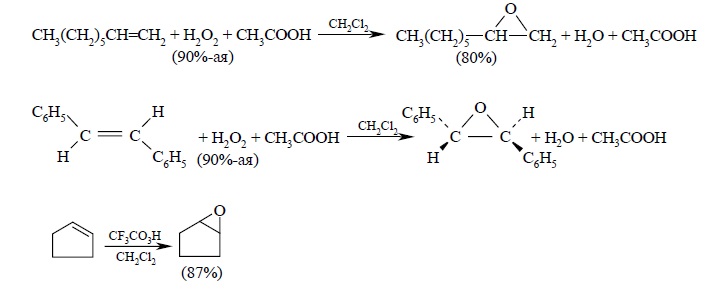

Алкены, с двойной связью, сопряженной с карбонильной и карбоксильной группой или другим акцепторным заместителем, малоактивны, и для их окисления необходимо использовать более сильные окислители, такие как трифторперуксусную кислоту, получаемую из ангидрида трифторуксусной кислоты и 90%-й перекиси водорода в хлористом метилене. Альтернативный метод эпоксидирования заключается во взаимодействии алкена с нитрилом и 90%-й перекисью водорода: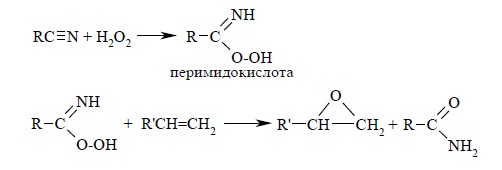

Простейший оксиран – окись этилена – получают в промышленности окислением этилена кислородом в присутствии серебра как катализатора: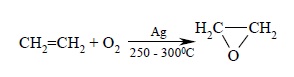
Трехчленное кольцо оксиранов легко раскрывается под действием самых разнообразных нуклеофильных реагентов. Эти реакции подробно будут обсуждаться в главе 11, посвященной ациклическим и циклическим простым эфирам. Здесь же будет рассмотрен только гидролиз эпоксидов. Гидролиз эпоксидов катализируется как кислотами, так и основаниями. В обоих случаях образуются вицинальные диолы, т.е. гликоли. При кислотном катализе в первой стадии происходит протонирование атома кислорода эпоксида с образованием циклического оксониевого иона, который раскрывается в результате нуклеофильной атаки молекулы воды: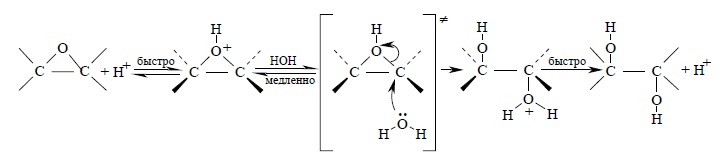

Ключевой стадией в раскрытии кольца, определяющей скорость всего процесса, является нуклеофильная атака водой на протонированную форму эпоксида. С точки зрения механизма этот процесс аналогичен раскрытию бромониевого иона при нуклеофильной атаке бромид-иона или другого нуклеофильного агента. С этих позиций стереохимическим результатом должно быть образование транс-гликолей при расщеплении циклических эпоксидов. Действительно, при кислотно-катализируемом гидролизе циклогексеноксида или циклопентеноксида образуются исключительно транс-1,2-диолы: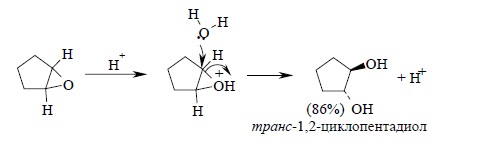

Таким образом, двухстадийный процесс эпоксидирования алкена с последующим кислотным гидролизом эпоксида суммарно соответствует реакции анти-гидроксилирования алкенов.
Обе стадии анти-гидроксилирования алкенов можно совместить, если алкен обрабатывать водной 30 – 70%-й перекисью водорода в муравьиной или трифторуксусной кислоте. Обе эти кислоты являются достаточно сильными для того, чтобы вызвать раскрытие эпоксидного цикла, поэтому их обычно используют для анти-гидроксилирования алкенов, например: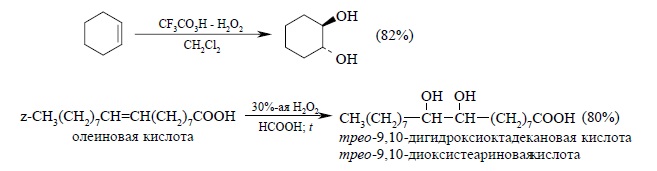

Раскрытие эпоксидного кольца, катализируемое основанием, также приводит к образованию транс-гликолей: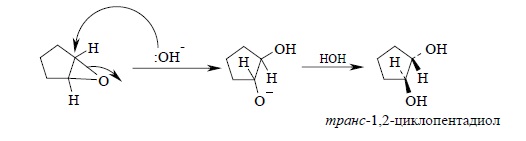

Следовательно, двухстадийный процесс эпоксидирования алкенов с последующим щелочным гидролизом эпоксидов также является реакцией анти-гидроксилирования алкенов.
Третий современный метод анти-гидроксилирования алкенов был предложен и разработан К. Прево (1933 г.). Алкен нагревают с йодом и бензоатом или ацетатом серебра в безводном бензоле или CCl4. транс-Присоединение к двойной связи первоначально приводит к образованию йодэфира, в котором йод далее замещается бензоат-ионом, и получается дибензоат гликоля: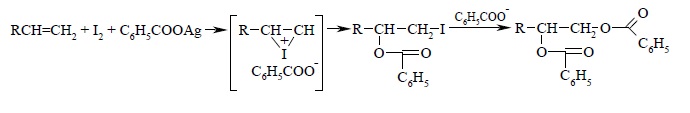

Реакция Прево в безводной среде приводит к образованию того же диола, что и эпоксидирование алкенов с последующим гидролизом: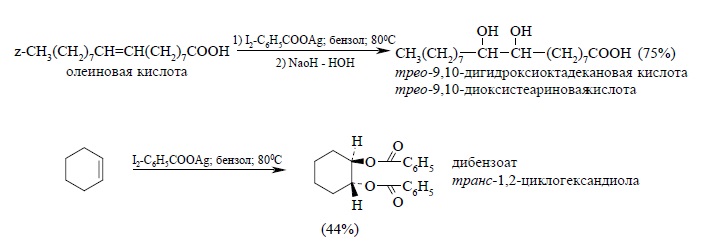

Таким образом, реакция Прево представляет собой более дорогостоящую модификацию других методов анти-гидроксилирования алкенов. Однако для чувствительных к действию кислот соединений этот метод имеет очевидные преимущества перед методом анти-гидроксилирования с помощью перкислот и последующего кислотного гидролиза эпоксида.
Некоторые соли и оксиды переходных металлов высших степенях окисления являются эффективными реагентами син-гидроксилирования двойной связи. Окисление алкенов перманганатом калия – один из старейших методов син-гидроксилирования двойной связи – продолжает широко использоваться несмотря на свойственные ему ограничения. цис-1,2-Циклогександиол был впервые получен В.В. Марковниковым еще в 1878 г. гидроксилированием циклогексена водным раствором перманганата калия при 0ºС: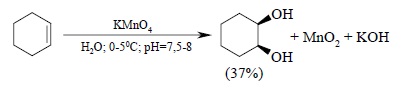

Этот метод в дальнейшем получил развитие в работах русского ученого Е.Е, Вагнера, поэтому син-гидроксилирование под действием водного раствора перманганата калия носит название реакции Вагнера. Перманганат калия является сильным окислителем, способным е только гидроксилировать двойную связь, но и расщеплять образующийся вицинальный диол. Чтобы по возможности избежать дальнейшего расщепления гликолей, необходимо тщательно контролировать условия реакции. Наилучшие результаты достигаются при гидроксилировании алкенов в слабощелочной среде (pH ~ 8) при 0 – 5ºС разбавленным ~ 1% водным раствором KMnO4. Тем не менее выходы гликолей обычно невелики (30 – 60%):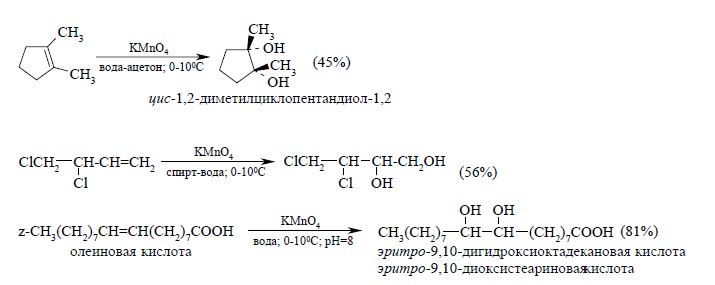

Первоначально при окислении алкенов перманганатом калия образуется циклический эфир марганцевой кислоты, который немедленно гидролизуется до вицинального диола: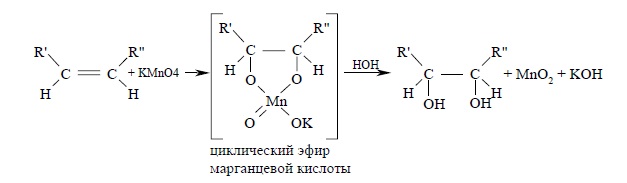

Циклический эфир марганцевой кислоты как интермедиат никогда не был выделен, однако его образование следует из экспериментов с меченым 18O перманганатом калия. К. Вайберг с сотрудниками (1957 г.) показали, что оба атома кислорода в гликоле оказываются мечеными при окислении алкена KMn18O4. Это означает, что оба атома кислорода переходят от окислителя, а не из растворителя – воды, что находится в хорошем соответствии с предлагаемым механизмом.
Другой метод син-гидроксилирования алкенов под действием оксида осмия (VIII) OsO4 был предложен Р. Криге в 1936 г. Тетраоксид осмия представляет собой бесцветное кристаллическое вещество, хорошо растворимое в эфире, диоксане, пиридине и других органических растворителях. При взаимодействии тетраоксида осмия с алкенами в эфире или диоксане образуется черный осадок циклического эфира осмиевой кислоты – осмат, который легко может быть изолирован в индивидуальном виде. Присоединение OsO4 к двойной связи заметно ускоряется в растворе пиридина. Разложение осматов до вицинальных диолов достигается действием водного раствора гидросульфита натрия или сероводородом: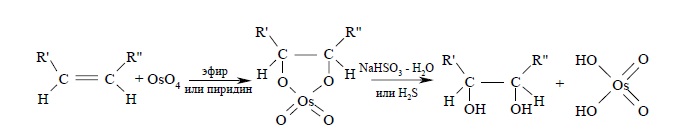

Выходы продуктов син-гидроксилирования алкенов в этом методе значительно выше, чем при использовании перманганата в качестве окислителя. Важным достоинством метода Криге является отсутствие продуктов окислительного расщепления алкенов, характерного для перманганатного окисления: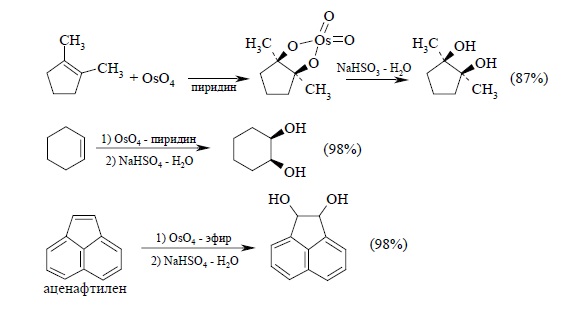

Тетраоксид осмия – дорогой и труднодоступный реагент, к тому же он очень токсичен. Поэтому оксид осмия (VIII) используют для синтеза малых количеств труднодоступных веществ с целью получения наиболее высокого выхода диола. Для упрощения син-гидроксилирования алкенов под действием OsO4 была разработана методика, позволяющая использовать лишь каталитические количества этого реагента. Гидроксилирование осуществляется с помощью перекиси водорода в присутствии OsO4, например: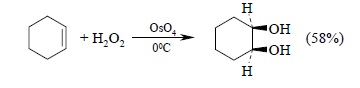

Интересно отметить, что высшие оксиды других переходных металлов (V2O5, WO3, MoO3 и др.) катализируют анти-гидроксилирование алкенов.
Р. Вудворд в 1958 г. предложил альтернативный трехстадийный способ син-гидроксилирования алкенов. Первоначально алкен превращают в транс-йодацетат в результате взаимодействия с йодом и ацетатом серебра в уксусной кислоте. Затем галоген замещаю на оксигрупу при обработке водной уксусной кислотой при нагревании. Последняя стадия заключается в гидролитическом отщеплении ацетатной группы: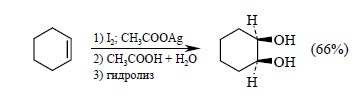

В заключение этого раздела приведем стереохимические отношения между алкеном цис- или транс-конфигурации и конфигурацией образующегося вицинального гликоля, который может быть цис- или транс-изомером, эритро- или трео-формой, мезо- или d-,l-формой, в зависимости от заместителей в алкене: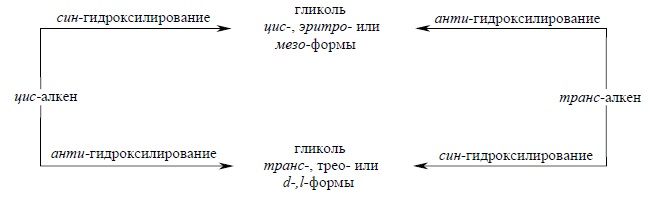

Аналогичные стереохимические отношения наблюдаются и в других реакциях син- или анти-присоединения водорода, галогеноводородов, воды, галогенов, гидридов бора и других реагентов по кратной связи.